Conclusiones del Grupo de Trabajo sobre biosimilares.
|
Marcos Domínguez. Madrid
Están entre nosotros desde el año 2006 y sin embargo es ahora cuando se encuentran en boca de todo el mundo. La combinación de crisis económica y vencimientos de patente de medicamentos complejos ampliamente utilizados ha puesto en guardia a laboratorios, autoridades, farmacéuticos, médicos y pacientes. Todos están expectantes ante la llegada de los nuevos biosimilares.

Francisco Zaragozá, vocal de Investigación y Docencia del Cgcof.
|
El pasado septiembre, la Comisión Europea autorizó dos biosimilares de infliximab. Era la primera vez que se aprobaba la comercialización de un anticuerpo monoclonal biosimilar, algo completamente distinto a los hasta ahora existentes. Francisco Zaragozá, catedrático de Farmacología y vocal de Investigación y Docencia del Consejo General de Colegios de Farmacéuticos (Cgcof), explica que “tenemos que distinguir entre al menos dos tipos de biosimilares”. Unos, los aprobados hasta ahora, “son obtenidos por técnicas de ADN recombinante a imagen de una hormona o de un factor trófico”, moléculas muy conocidas y con un mecanismo de acción claro y conocido. Por ejemplo, la hormona del crecimiento: “Cuando hay carencia de esa hormona, se sustituye y ya está”.
Sin embargo, “con los anticuerpos monoclonales el mecanismo de acción solo es parcialmente conocido”. Además, las técnicas para producir un biosimilar “son distintas por definición” a las de un medicamento innovador, por lo que pueden producirse diferencias. Por tanto, “hay que ser cautos con la intercambiabilidad”.

Javier Cortés, especialista de la SEOM.
|
La palabra intercambiabilidad, junto con otra, sustitución, están en el centro de la polémica de estos fármacos. La primera implica que innovador y biosimilar puedan usarse para una indicacación concreta como equivalentes terapéuticos como un procedimiento clínico más. La segunda, que la decisión de usar uno u otro esté fuera del ámbito clínico.
Siempre por decisión del médico
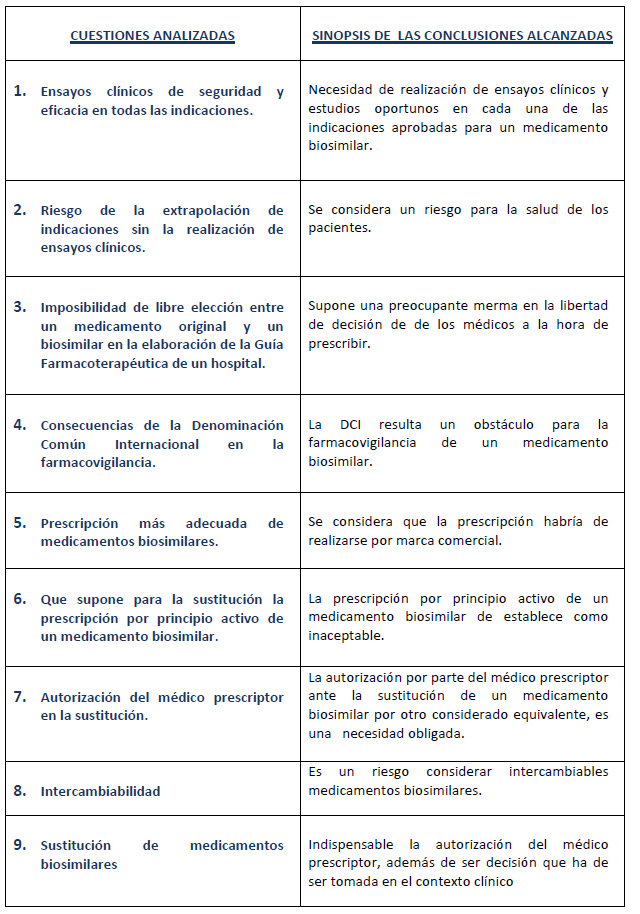
Por todo lo anterior, Javier Cortés, del servicio de Oncología Médica del Hospital Vall d’Hebron y especialista de la Sociedad Española de Oncología Médica (SEOM), señala que “la sustitución automática debe estar siempre prohibida”. Debe ser siempre decisión del médico prescriptor. La Agencia Europea del Medicamento (EMA) exige unos estándares de seguridad y eficacia que alivia el temor de que aparezcan “biochapuzas”, pero no por ello está de acuerdo en que se aprueben indicaciones para las que no ha habido un ensayo específico, basándose en la extrapolación. “En general, la extrapolación ha de evitarse”.

Fernando Carballo, presidente de la SEPD.
|
(1).jpg)
Teresa Tejerina, presidenta de la SEF.
|
Cuando se aprobaron los dos biosimilares de infliximab hubo expertos y sociedades científicas que se manifestaron contrarias a su uso en indicaciones extrapoladas. Los ensayos en los que se basó la EMA para su aprobación evaluaban su acción en artritis reumatoide, pero al estar indicado el fármaco de referencia para enfermedades inflamatorias intestinales, se hizo lo mismo para su biosimilar. Para Fernando Carballo, presidente de la Sociedad Española de Patologías Digestivas (SPED), se trata de un error. “En el actual escenario es más necesario que nunca que la eventual experiencia que pueda tenerse con los biosimilares en la enfermedad inflamatoria intestinal esté sujeta a estrictos criterios de registro y seguimiento dentro de un estrecho plan de farmacovigilancia”.
Nombre comercial vs DCI
Para asegurar esta farmacovigilancia, otro de los puntos que exigen los expertos es que la prescripción del médico se haga por nombre comercial y no por su denominación común, algo que se recomienda desde Europa. “La denominación común resulta un obstáculo para la farmacovigilancia de un medicamento biosimilar” señala Teresa Tejerina, presidenta de la Sociedad Española de Farmacología (SEF), “y el numero de lote y fabricante”, que en principio puede asegurar la trazabilidad, “no se han planteado nunca como referentes”. Por tanto, “la prescripción por principio activo de un medicamento biosimilar es inaceptable”.

Alejandro Toledo, presidente de la AGP.
|
.jpg)
Manuel Castaño, vicepresidente de la SER.
|
La Ley 10/2013, de reforma de la de Garantías y Uso Racional de los Medicamentos, eliminó una polémica frase que incluía el RD 16/2012 sobre la posible sustitución de un medicamento por su “biosimilar correspondiente". Sin embargo, los pacientes, principales afectados, siguen temiendo que la actual estrechez económica fuerce a las administraciones a colocar el criterio económico por encima del clínico. “Es uno de nuestros principales temores”, manifiesta Alejandro Toledo, presidente de la Alianza General de Pacientes (AGP). Señala que no es la normativa europea la que les preocupa sino “cómo está redactada la legislación que permite el intercambio de fármacos sin la autorización del especialista y motivado exclusivamente por criterios económicos”. Por ello, sostiene que “el paciente debería ser informado y poder elegir libremente”.
La Alianza General de Pacientes formó un grupo de trabajo de expertos, que elaboró un informe sobre biosimilares donde se indicaba la necesidad de una prescripción por nombre comercial, la intercambiabilidad siempre a criterio del médico prescriptor y la realización de ensayos clínicos para cada una de las indicaciones para las que se apruebe el fármaco. Concluyendo, ¿debería el biosimilar ser tratado como un medicamento innovador más? Manuel Castaño, vicepresidente de la Sociedad Española de Reumatología (SER) y miembro de ese grupo de trabajo, afirma que “deben ser tratados como un fármaco más”, matizando que, “al tratarse de medicamentos nuevos, requieren un seguimiento adicional tanto por parte de las autoridades sanitarias como por los profesionales para obtener más información sobre su perfil de seguridad en la práctica clínica diaria”.
|
